Research Interests
I am broadly interested in the application of computational methods, data science, and machine learning to chemical and biological problems. At Merck, my work focuses on the design of new experimental methods for the collection of large chemical datasets, the development of a microfluidic platform for ultra-high-throughput protein engineering, and the study of reaction mechanisms through quantum chemistry and kinetic isotope effects. Below, I summarize some completed projects.
Screening for Generality in Asymmetric Catalysis
When I was a graduate student in Dave Evans’ group, one of my colleagues quipped that “the only good model substrate is the enantiomer.” The phenomenon that small variations in structure can have a big impact on reactivity and selectivity is widely appreciated, but why is it that when we develop asymmetric reactions, we almost always use a single model substrate? The simple answer is that, until now, it would be too much work to design, setup, and assay multiple substrates at once. Advances in computational methods and high-throughput experimentation (HTE) have made the first two challenges much easier, but chiral analysis remains slow.
Many efforts have focused on speeding up chiral separations (faster chromatography or using plate readers), but what if you could get more out of each measurement? We propose a scheme where you run reactions normally in separate vessels, but then combine aliquots into a single chromatography vial for analysis. As long as the products have different masses, the enantioselectivities can be read out from the ion chromatograms:
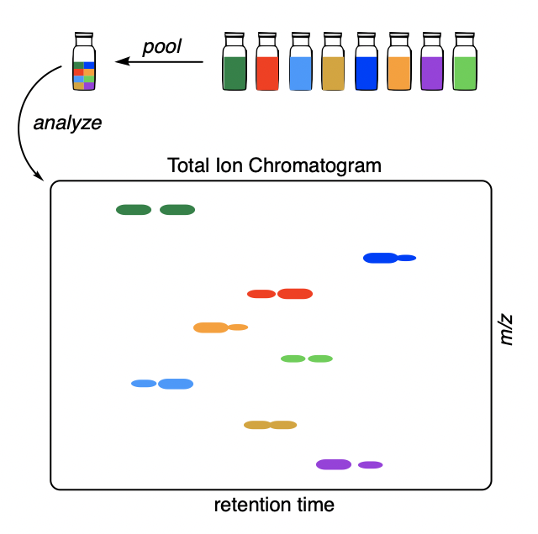
This is essentially a trick that uses existing equipment and a bit of software to squeeze the most out of every measurement. Because enantiomers have identical response factors, no calibration curves are necessary! This method really can be used “out of the box.” In fact, we have already measured thousands of enantioselectivities this way (not all of it is published). In the paper, we used the method to find a general catalyst system for the Pictet–Spengler reaction:
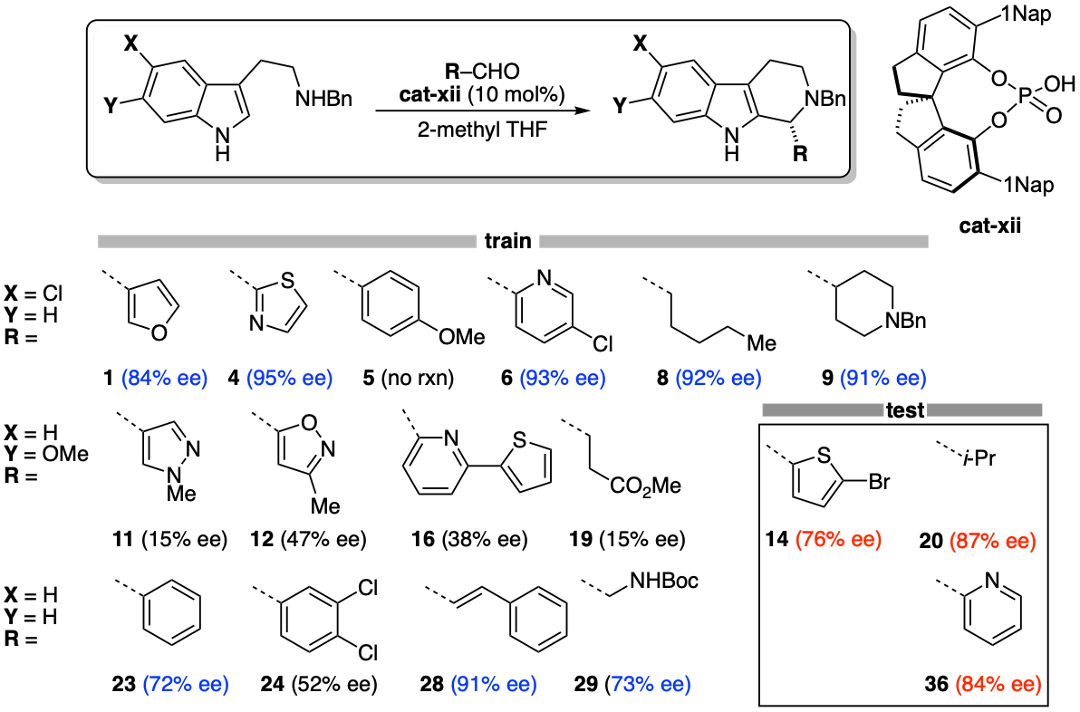
As you might expect, there is no single model substrate that is representative, and multiple substrates are needed to find a solution with broad scope. For example, we found that while toluene was the optimal solvent for a canonical model substrate, it was actually much worse for most substrates! After optimizing conditions across a training set of 14 substrates, we found that SPINOL xii was the most general. Amazingly, the selectivities were good across a test set of 3 substrates! It is very unusual to find good enantioselectivity across a range of aliphatic, aromatic, and heteroaromatic substrates. This result gives me a lot of hope that there are many more general solutions waiting to be found!
Publication: Nature 2022 (in press)
read the article another non-technical summary
Nucleophilic Aromatic Substitutions
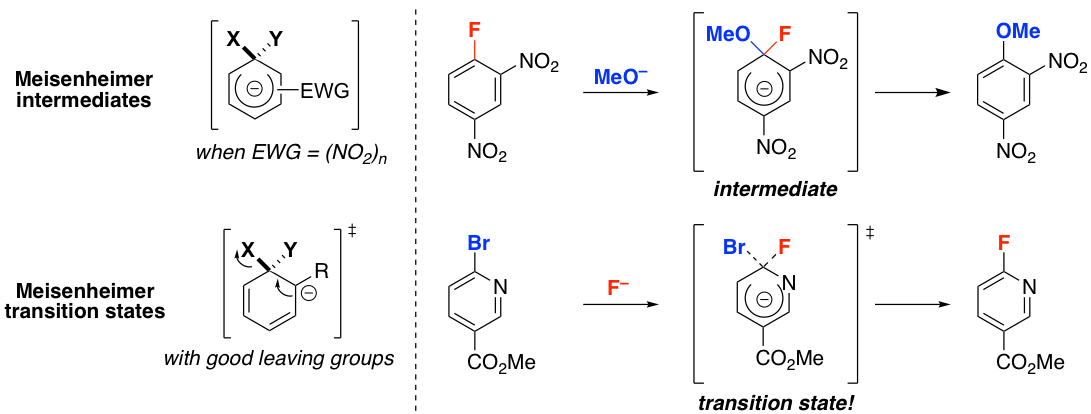
Nucleophilic aromatic substitution (SNAr) is one of the most frequently used reaction classes in medicinal chemistry. For over 60 years, the accepted mechanism has involved the stepwise addition of a nucleophile to generate a Meisenheimer intermediate, followed by leaving group elimination. Using kinetic isotope effect measurements (please see below), we have shown that prototypical SNAr reactions can occur in a single concerted step. Computations indicate that anionic Meisenheimer complexes are only stable when they are stabilized by strong electron withdrawing groups and no good leaving groups are present. These requirements are not met by the heteroaryl substrates typically used in the production of pharmaceuticals. In fact, our calculations indicate that most SNAr reactions are concerted.
Publication: Nature Chem. 2018, 10, 917. Read the blog post here.
Ultrasensitive Measurement of Kinetic Isotope Effects
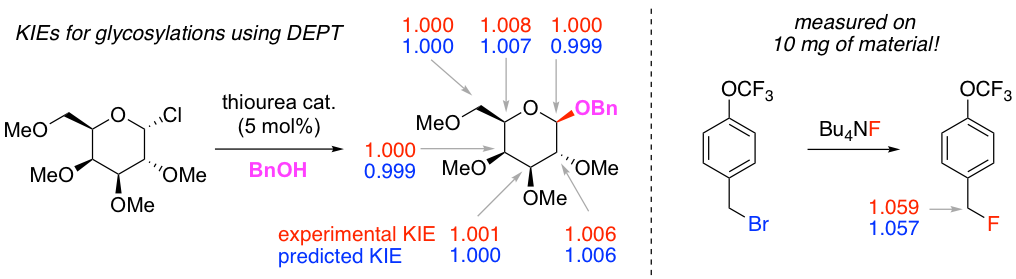
Kinetic isotope effects (KIEs) are a powerful tool for studying reaction mechanisms. Traditionally, the measurement of 12C/13C KIEs requires either the laborious synthesis of isotopically labeled material or long NMR acquisition times on large amounts of natural abundance material. We have developed two natural abundance methods for measuring KIEs that drastically reduce the amount of time and material required. One method uses distortionless enhancement by polarization transfer (DEPT) to reduce the acquisition time by an order of magnitude over that which is normally required. This is a general method that is already being used to study a variety of reactions by other groups.
An even more powerful method applies when fluorine is adjacent to the site of interest. Our strategy is to quantitate the 13C satellites in 19F spectra in which the large 12C–19F resonances have been suppressed. This reduces the time required by two orders of magnitude, allowing KIEs to be measured on tiny quantities of material (e.g., 10 mg in one overnight acquisition). This method will be useful for studying both fluorination and defluorination reactions, which are of considerable synthetic interest. We have applied this method to the study of nucleophilic aromatic substitution reactions (see above).
We have also used the DEPT method to study a recently developed catalytic glycosylation reaction. In general, glycosylations occupy the boundary between the SN1 and SN2 mechanisms because the cationic intermediate (i.e., oxocarbenium ion) is relatively stable. The KIE measurements are quantitatively consistent with theoretical predictions, which show that the role of the catalyst is to effect a stereospecific SN2 displacement. This not only explains the method’s broad substrate scope, but also provides insight into how the catalyst achieves dual activation of both the nucleophile and electrophile. These findings will have important implications for the design of future glycosylation catalysts.
Publications: Science 2017 355 162; J. Am. Chem. Soc. 2017 139 43.
NMR Chemical Shift Prediction
When NMR spectroscopy was introduced in the 1960s, it revolutionized organic chemistry by enabling the routine determination of molecular structure. One reason NMR spectroscopy is so useful is that chemical shifts are exquisitely sensitive to small changes in molecular environment. As a result, it is possible to use predicted chemical shifts (from DFT calculations) to distinguish between possible structures. However, conventional predictions ignore molecular vibrations and rotations, instead treating molecules as static structures. We showed that this leads to systematic errors in predicted NMR shifts that must normally be corrected by empirical scaling factors. When motions are accounted for (through quasiclassical dynamics simulations), these systematic errors disappear.
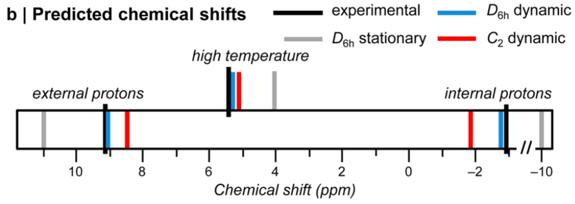
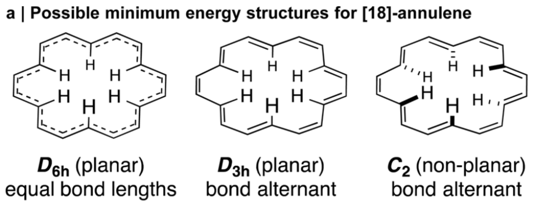
Our method leads to the most accurate predictions for both gas-phase absolute shieldings and solution-phase chemical shifts presently available. We are also able to treat systems with unusual dynamics that give very poor results with scaling factors that were developed for typical molecules. For example, the scaled prediction for the internal protons of the Hückel aromatic [18]-annulene are some 7 ppm in error! (0.3 ppm is typical.) The discrepancy is due to facile out-of-plane motions that partially break aromaticity. In other words, the dynamic structure of [18]-annulene is considerably less conjugated than its stationary structure would otherwise suggest. As a result, the stationary predictions are qualitatively incorrect and there is a pronounced temperature dependence on chemical shift. Our prediction method reproduces these effects quantitatively. This represents an important step towards achieving chemical accuracy in magnetic shielding predictions, which will not only be useful for studying exotic structures, but also elucidating the structures of natural products, the characterization of new materials, and determining the structures of proteins.
Publication: J. Chem. Theor. Comput. 2015, 11, 5083